Projects
In the Firat-Karalar lab, we study the biology of the mammalian centrosome/cilium complex, with a specific focus on how these structures are assembled, maintained and dynamically altered in response to signals and what goes awry in disease like cancer and ciliopathies. To this end, we ask clear scientific questions and address them in cell lines and in vitro tracheal cultures using a combination of genome editing, proteomics, cell biology and biochemistry approaches. When required, we learn new techniques by attending workshops, consulting the experts and initiating collaborations with other labs. In the long term, we aim to understand how and why cells compartmentalize through membrane-less structures and reveal the molecular and cellular defects underlying cancer and ciliopathies.
Research Projects
The centrosome is the main microtobule-organizing center of animal cells that plays important roles in various cellular processes including the formation of cilia and flagella. At the core of the centrosome are two centrioles surrounded by pericentriolar material that nucleates and organizes microtubules. In addition to these structures, most cells have an array of granules, called centriolar satellites, tha localize around the centrosome. Importantly, there are many links between the centrosome/cilium complex and human disease. Structural and numerical abnormalities of the centrosome have long been associated with cancer and also mutations affecting components of the centrosome and cilium cause human genetic diseases including ciliopathies and primary microcephaly. The central questions behind our work is understanding how centrosomes and cilia are assembled, maintained and dynamically altered during cell cycle, as well as elucidating what goes awry in diseases associated withabnormalities in centrosome/cilium complex. Examples to diseases are cancer and ciliopathies that are characterized by primary microcephaly, cystic kidney disease, retinal degeneration and obesity.
1- Dissecting the structure and function of centriolar satellites as key regulators of the mammalian centrosome/cilium complex (ERC project)
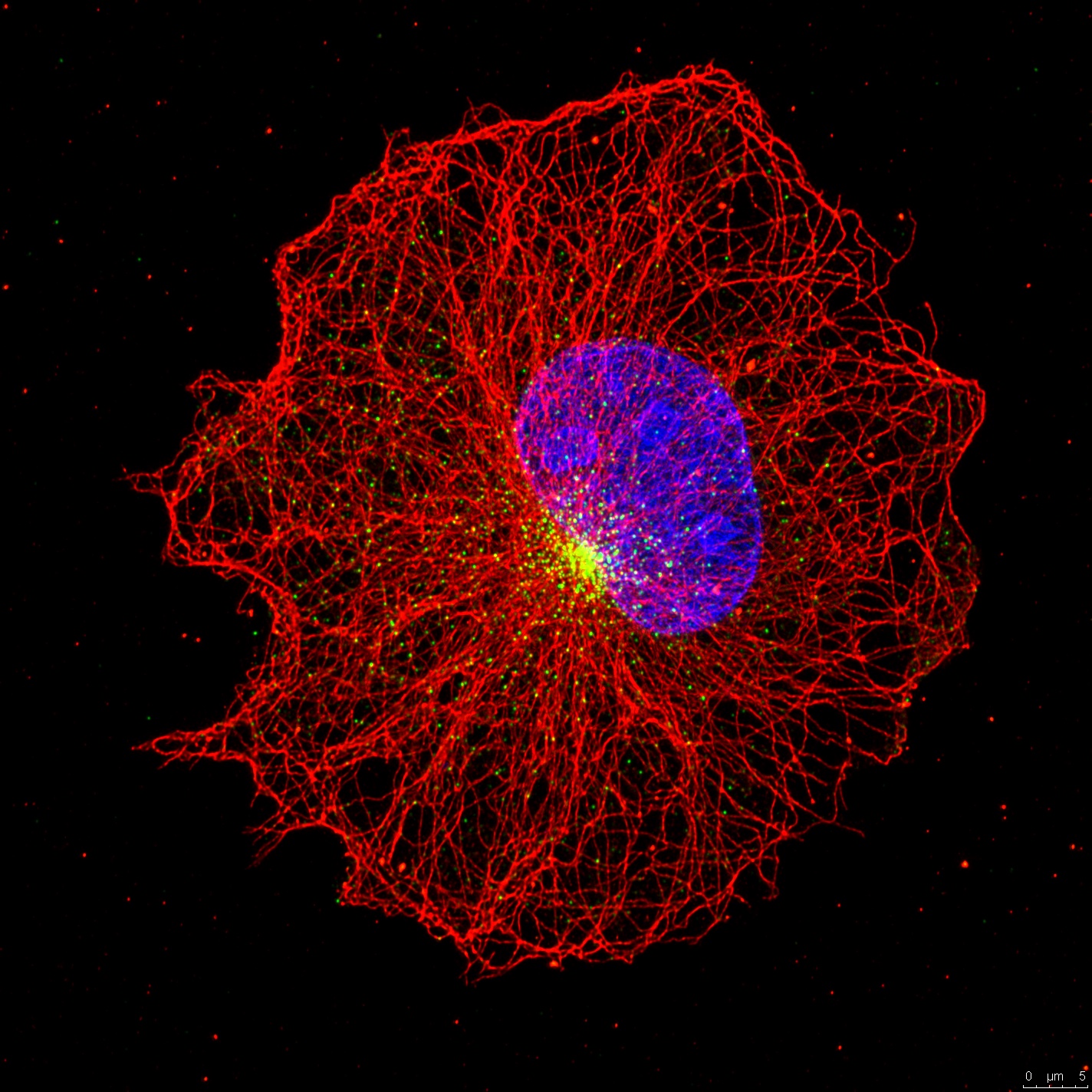
Centriolar satellites are the array of granules that localize around the centrosome/cilium complex in mammalian cells. Only recently interest in the satellites has grown because mutations affecting satellite components were shown to cause ciliopathies, microcephaly and schizophrenia. Remarkably, many centrosome/cilium proteins localize to these structures and we lack understanding of when, why and how these proteins localize to satellites. To address these questions, we aim to identify the nature of regulatory and molecular relationship between satellites and the centrosome/cilium complex, .elucidate the role of satellites in proteostasis of centrosome/cilium proteins and investigate the functional significance of satellite-localization of centrosome/cilium proteins during processes that go awry in human disease. Using a multidisciplinary approach, the results of these studies will expand our knowledge of the spatiotemporal regulation of the centrosome/cilium complex and provide new insights into pathogenesis of ciliopathies and primary microcephaly.
2- Application of novel proximity labeling approaches to generate spatial and temporal interaction maps for the centrosome, cilia and satellites (Newton Fund, EMBO Installation Grant)

In previous work, we combined BioID proximity labeling approach with centrosome enrichments to identify the proteins at the centriole origin of duplication. The results of this study demonstrated that the BioID approach is superior in defining temporal and spatial interactions among centrosome duplication proteins over standard biochemical approaches. To generate a proximity interaction map among centrosome proteins, we aim to apply recently developed proximity labeling approaches with higher temporal resolution to other parts of the centrosome including centriolar satellites, distal and sub-distal appendages and pericentriolar material. The results of these studies will be significant in elucidating how centrosomes are assembled, maintained and dynamically altered during the cell cycle.
3- Dissecting the mechanisms underlying centriole amplification and cilia formation in multiciliated cells
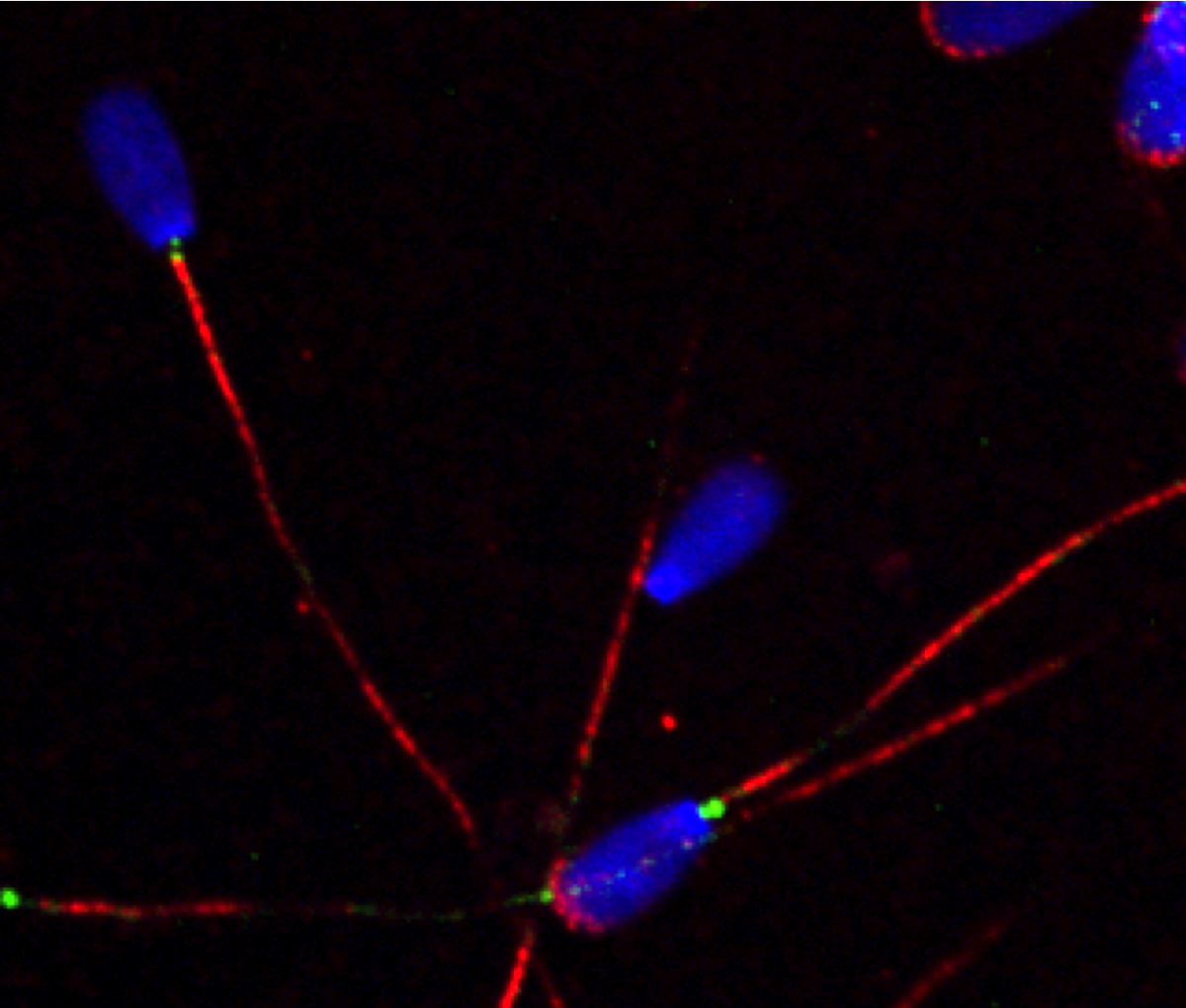
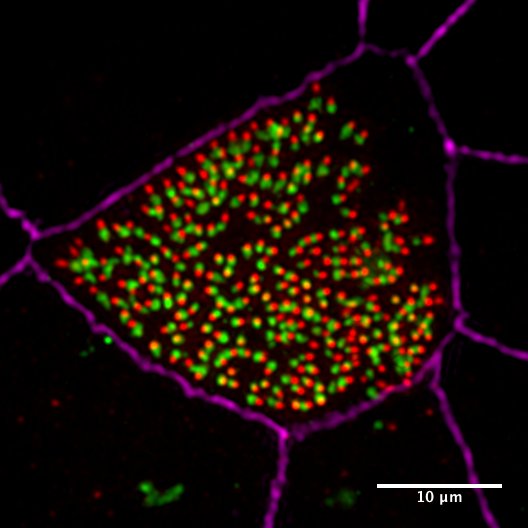
In previous work, we developed a biochemical purificaiton protocol of centrioles from sperm cells and identified the sperm centriole proteome using mass spectrometry. The sperm centriole proteome has revealed many previously uncharacterized centrosome proteins, many of which are conserved in the multiciliated epithelial cells. To elucidate the molecular mechanism of how hundreds of centrioles and cilia assemble in multiciliated epithelial cells, we use in vitro multiciliated mouse tracheal cultures as a model system and define the key proteins required for these processes using a combination of cell biology and proteomics approaches.
4- Elucidating the molecular defects underlying ciliopathies

Primary cilium functions as a nexus for important signaling pathways including Hedgehog signaling. Defects associated with primary cilium structure and function cause multi systemic human genetic diseases known as ciliopathies, which are characterized by primary microcephaly, retinal degeneration, obesity and cystic kidney disease. Over the years there has been a significant number of genes identified that cause ciliopathies when mutated. However, the genotype-phenotype relationship for these genes are not known. Our goal is to elucidate the molecular defects that caue ciliopathies. To this end, we collaborate with cilinical geneticists and thus work with patient samples to identify the functional defects in these cells.